A brighter future for the human race relies heavily on energy efficient electronics and renewable energy systems. With an upper efficiency of around 23% so far, conventional Silicon based solar cells wouldn’t help curb the growing electricity demands in the next generation. Further, Light Emitting Diodes (LED) need to be made more luminous for wider applications. A unique class of materials called ‘Perovskites’ which caught the eyes of scientists a decade ago might hold potential revolutionary secrets for future optoelectronics devices.
Materials, whose crystal structure resembles that of a Calcium Titanate (CaTiO3) crystal are called Perovskites. ABX3 is the general formula for such materials, is also referred to as the ‘Perovskite Structure’. Here A & B are cations, like Calcium & Titanium in CaTiO3 and X is an anion, like Oxygen. Generally, oxygen and halogens are considered as anions in double perovskites. For constructing photovoltaic application devices, the employed material should be structurally stable and its optimum band gap should range within 1eV & 2eV (eV or electron volts is a unit of energy used for subatomic particles). Unlike Silicon, which is highly stable and resembles a diamond structurally, halide single perovskites (ABX3) are unstable.
In this pursuit, ‘Double Perovskites’ with a structural formula A2B’B’’X6 looks interesting, as they are exceptionally stable in contrast to the single perovskites. Here, A, B’ and B’’ are all cations while X is an anion. Double perovskites are nothing but the three dimensional extension of the single perovskite (ABX3) structure. In comparison to single perovskites, these double perovskites exhibit a larger band gap (greater than 2eV) which pose a hurdle. Can we tune these double perovskites catering to our needs for novel optoelectronics applications? Is it possible to modify and tweak them by tailoring their band gap in the required optimum range?
Recent experimental research from the Multifunctional Materials Laboratory (MFML) in the Department of Physics at IIT Madras shows that it is possible to tune the band gap of these semiconductor materials. “For the first time, we showed the band gap tunability in these double perovskites, with our study carried out on Cs2Ag1-xNaxBiCl6 double perovskite material” said Prof. Sudakar Chandran, Associate Professor and head, MFML lab in the Department of Physics, IIT Madras, who guided the study.
Cs2Ag1-xNaxBiCl6 is the double perovskite made of Caesium (Cs), Silver (Ag), Sodium (Na) and Bismuth (Bi) as cations, and Chlorine (Cl) as anion. Prof. Chandran with his student Mr. Athrey C. Dakshinamurthy synthesized intermediate Cs2Ag1-xNaxBiCl6 double perovskite alloys by mixing Silver and Sodium at B’ cation, and varying the elemental amount via the parameter x, valued within 0 and 1. At x = 0 end, we obtain the Cs2AgBiCl6 double perovskite and at x = 1 end, the Cs2NaBiCl6 double perovskite is formed.


“Varying the concentrations of Silver and Sodium, alters the way the band gap changes in Cs2Ag1-xNaxBiCl6.This novel way of altering the band gap can be employed as a tool for tweaking the electronic and optical properties of these double perovskites” pointed Prof. Chandran. Band gap is an energy difference, which the electrons of a semiconductor material should acquire in order to conduct electricity. In Solar Cells, electrons acquire such energy via sunlight.
“More intriguingly, this double perovskite alloy showed a band gap bowing effect” Prof. Chandran mentioned. What is this band gap bowing effect? Band gap of double perovskite materials like Cs2Ag1-xNaxBiCl6 varies nonlinearly upon varying the concentration of various cations. This nonlinear behavior is called the band gap bowing effect. But, how does this nonlinearity emerge in the band gap?
Variations in the bonding characteristics of double perovskites happen by varying the amount of elements used in their formation. This creates local distortions in the double perovskite’s structure. Also, various chemical effects arise due to the intermixing of atomic orbitals – an electron’s most probable house in an atom. Due to the varying the concentration of these elements (like of Sodium & Silver here)such chemical changes are iduced. All of this gives rise to what we observe experimentally as the band gap bowing effect.
“It is for the first time that such sublattice distortion is found to be the reason behind the band gap bowing effect in a double perovskite. We have shown that via our experiments on Cs2Ag1-xNaxBiCl6” remarked Prof. Chandran.
Praising the work carried out by Prof. Chandran and team, Prof. Amlan J. Pal of the Indian Association for the Cultivation of Science (IACS), Kolkata and a renowned expert in the field said “The results that authors have observed and reported are truly interesting, especially the expansion of double perovskite lattice. The expansion takes place in spite of the increasing concentration of the Sodium cations, which have smaller effective ionic radius than the Silver ones. This rightfully infers a significant and dominant role of structural alterations behind the observed bowing phenomenon.”
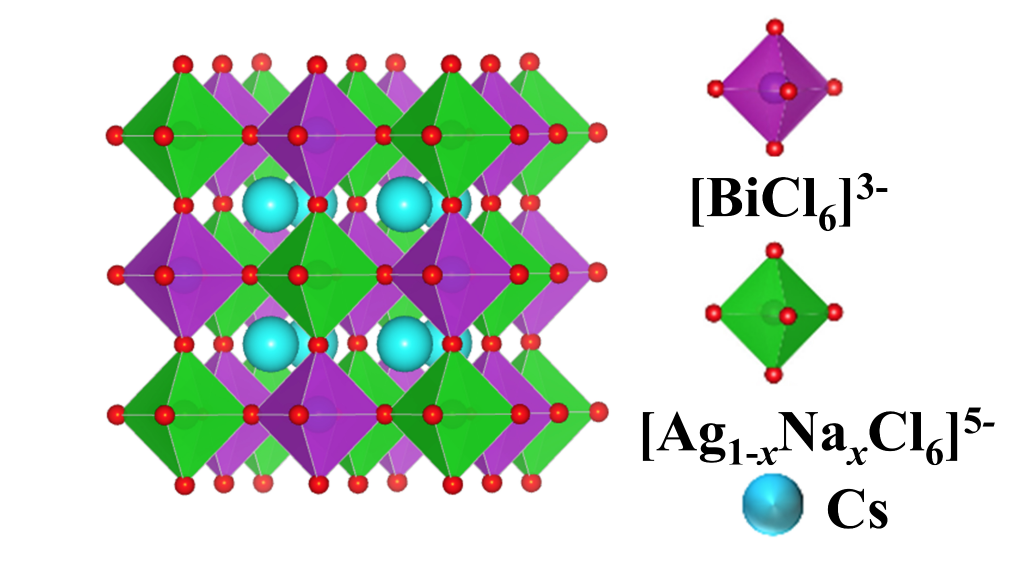
Making use of X-ray diffraction methods, Prof. Chandran’s group observed that their double perovskite exhibits a rock salt ordering of Silver & Bismuthcations. Silver possesses one unit of positive charge on it while Bismuth holds a positive charge of three. Their double perovskite material consists of a three dimensional framework of corner connected [BiCl6] anion and [Ag(Na)Cl6] anion octahedra, with Caesium cations occupying the cuboctahedral cavities (see Figure 1). [BiCl6] anion holds a negative three charge on it while [Ag(Na)Cl6] anion holds a negative five charge on it.
How did Prof. Chandran and his student observe the distortions in a double perovskite alloy? Raman Spectroscopy is the tool they used to look into their Cs2Ag1-xNaxBiCl6 samples. With the help of Raman Spectroscopy, we can infer the local distortions in any material as well as the bonding and arrangements of the atoms in the respective constituent molecules. Strong interplay between the [BiCl6] anion octahedra and the [Ag(Na)Cl6] anion octahedra was observed via these methods, which results in the sublattice distortion of the Cs2Ag1-xNaxBiCl6 double perovskite, and thereby imparting a nonlinearity in the band gap change.
Commending the study, Prof. Amlan added “An in depth Raman spectroscopic investigation by the authors revealed a strong correlation between the change in band gap and Raman spectroscopic observables like the Raman mode frequency shift, Raman peak width and the ratio of Raman modes’ intensities. These observations clearly suggest that the local distortions of [BiCl6] anion octahedra and the [Ag(Na)Cl6] anion octahedra impart a non-linearity in the change in the band gap.”
Crediting the splendid work by Prof. Chandran and his student, the journal, Applied Physics Letters has published this research work as an Editor’s Pick. The highlight of this work by the journal reads “Lead free double perovskites are gaining significant attention owing to their intriguing optical properties which will be useful for solar cell and light emitting diode applications. Sudakar group’s study unequivocally brings out the sub lattice distortion as a tool to engineer the electronic properties in these perovskites.”
Can we now construct more energy efficient solar cells and LEDs for a brighter future? We can! But we have to wait a while to see this happen. “Optimum band gap for photovoltaic applications is 1eV to 2eV, while optoelectronic devices have a wide range of band gap requirements. Our material was not within the range for photovoltaic application. But it is very much possible, by choosing the right composition, which we are currently working on” Prof. Chandran said.
Article by Mywish Anand
Here is the original link to the scientific paper:
https://aip.scitation.org/doi/10.1063/5.0046423